Identifizierung von Risikovarianten für das Restless Legs Syndrom
Forschungsbericht (importiert) 2007 - Max Planck Institut für Psychiatrie
Das Restless Legs Syndrom
Das Restless Legs Syndrom (RLS) gehört mit einer Häufigkeit von bis zu 10% in der Altersgruppe der über 65-Jährigen zu den wichtigsten neurologischen Erkrankungen. Frauen sind im Vergleich zu Männern doppelt so häufig betroffen. RLS-Patienten leiden an einem Bewegungsdrang der Beine, der meist mit sehr unangenehmen Missempfindungen verbunden ist. Die Symptome treten ausschließlich abends oder nachts in Ruhe- und Entspannungssituationen, z.B. beim Einschlafen, oder aus dem Schlaf heraus auf. Bewegung der betroffenen Extremitäten, wie beispielsweise Aufstehen und Umherlaufen, bessert die Beschwerden unmittelbar. Nach einer kurzen Ruhephase können die Symptome jedoch erneut auftreten. Bei mehr als 80% der RLS-Patienten zeigen sich im Wachen und im Schlaf außerdem sog. periodische Beinbewegungen (periodic leg movements, PLMs). Darunter versteht man stereotyp wiederholte Beugungen im Hüft-, Knie- und Fußknöchelgelenk, die vor allem im Non-REM-Schlaf auftreten. Diese Bewegungen können mit einer Weckreaktion im EEG einhergehen und zu einer Fragmentierung des Schlafs sowie zu verminderter Schlafeffizienz führen. Im weiteren Verlauf der Erkrankung kommen darüber hinaus schwere Ein- und Durchschlafstörungen vor. Eine Folge des RLS ist daher ein mangelnder Erholungseffekt des Schlafs, eine allgemeine Erschöpfung und verminderte Leistungsfähigkeit am Tage.
Sowohl die bisher vorliegenden Untersuchungen an RLS-Patienten mittels bildgebender und elektrophysiologischer Verfahren als auch das gute Ansprechen der Beschwerden auf eine dopaminerge und opioiderge Therapie sprachen für eine Entstehung der Erkrankung im zentralen Nervensystem. Eine Behandlung mit L-Dopa und Dopaminagonisten führt nämlich zur prompten Unterbrechung der sensiblen wie auch der motorischen Symptome. Schon in den ersten Beschreibungen des RLS wurde auf eine auffällige Häufung in den Familien der Betroffenen hingewiesen. Mehr als die Hälfte aller RLS-Patienten berichten über weitere erkrankte Familienangehörige. Zwillingsstudien zeigten eine Erblichkeit von 50%. Die genetische Komponente scheint daher bei der Entstehung der Erkrankung eine erhebliche Rolle zu spielen. Trotzdem konnten über viele Jahre keine DNS-Varianten identifiziert werden, die für das RLS ursächlich sind. Eine sorgfältige Phänotypisierung, d.h. eine genaue Symptombeschreibung aufgrund der klinischen Untersuchung der Patienten, stellt die wichtigste Voraussetzung für den Erfolg molekulargenetischer Studien zur Identifizierung genetischer Risikofaktoren dar. Durch den Vergleich der klinischen Charakteristika zwischen Patienten, die an der familiären und nicht-familiären Form der Erkrankung litten, konnte Juliane Winkelmann und ihre Arbeitsgruppe am MPI für Psychiatrie in München nachweisen, dass das familiäre RLS bereits in einem jüngeren Alter erstmals auftritt. Im Gegensatz dazu zeigen sich zwischen der familiären und nicht-familiären RLS-Form keine Unterschiede bezüglich der klinischen Symptomatik, wie z.B. den sensiblen oder motorischen Phänomenen, dem Verlauf der Erkrankung, der Lokalisation, den Schlafstörungen oder den polysomnographischen Parametern. Innerhalb einer Familie kann die Ausprägung der Beschwerden des RLS sehr unterschiedlich sein, d.h. mit schwer betroffenen Familienmitgliedern, die aufgrund der Symptome kaum noch schlafen können, und anderen, die nur an einzelnen Tagen im Jahr an leichten RLS-Symptomen leiden.
Unter der Annahme, dass das RLS durch ein dominantes Hauptgen vererbt wird, wurden genomweite Kopplungsuntersuchungen in großen RLS-Familien durchgeführt. Durch diese Studien identifizierten die Wissenschaftler mindestens fünf chromosomale Regionen, auf denen sich möglicherweise ein RLS-Gen befindet. Diese sog. RLS-Loci werden der Reihenfolge ihrer Beschreibung nach als RLS1-RLS5 bezeichnet und liegen auf den Chromosomen 12q, 14q, 9p, 20p und 2q, wobei ihre Identifizierung in RLS-Familien auf der Annahme eines rezessiven (RLS1) und eines dominanten Erbganges (RLS2-RLS5) beruhte. Eine kausale Sequenzvariante konnte durch diese Methode bisher allerdings nicht nachgewiesen werden. Die weite Verbreitung des RLS und seine genetische Heterogenität legen nahe, dass es sich um eine komplex-genetische Erkrankung handelt, bei der häufige genetische Varianten in unterschiedlichen Genen zur Entstehung der Erkrankung beitragen.
Genomweite Assoziationsstudien
Mit modernen Hochdurchsatzverfahren bei der Genotypisierung ist es möglich, eine große Anzahl von genetischen Varianten zu bestimmen. Eine der häufigsten Varianten sind sog. Einzelnukleotidpolymorphismen (single nucleotide polymorphism, SNPs). Es handelt sich hierbei um biallelische Marker, d.h., dass an einer bestimmten Stelle der DNS nur zwei der vier Basenpaare möglich sind. Über das gesamte menschliche Genom gemittelt tritt ca. alle 500 Basenpaare ein SNP auf, insgesamt mehrere Millionen. Bei genomweiten Assoziationsstudien werden eine große Anzahl SNPs, verteilt über das Genom, in einem Patienten- und Kontrollkollektiv ermittelt und ihre Häufigkeit (Allelfrequenz) durch statistische Verfahren verglichen. Eine unterschiedliche Allelfrequenz in beiden Populationen weist auf eine Assoziation mit dem entsprechenden Phänotyp hin. Um falsch-positive Assoziationen, z.B. durch systematische Fehler bei der Auswahl der Probanden, durch zufällige Besonderheiten in der Zusammensetzung der untersuchten Populationen oder durch Genotypisierungsfehler zu vermeiden, ist es unabdingbar, die Ergebnisse von genomweiten Assoziationsstudien in weiteren unabhängigen Populationen zu replizieren.
Die Identifizierung von genetischen Risikofaktoren
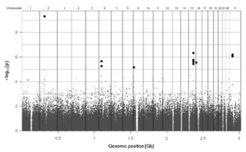
Die Studie wurde im Rahmen eines Kooperationsprojektes am Max-Planck-Institut für Psychiatrie und dem Institut für Humangenetik, Helmholtz-Zentrum München, Deutsches Zentrum für Gesundheit und Umwelt (Thomas Meitinger) durchgeführt. Durch den Einsatz von DNA-Chips der Firma Affymetrix wurden 500.000 häufige SNP-Varianten, verteilt über das gesamte menschliche Genom, in 400 RLS-Patienten und 1600 Kontroll-Probanden aus der Normalbevölkerung gemessen. Um eine möglichst homogene Population zu untersuchen, wurden ausschließlich RLS-Patienten mit einem familiären RLS in die Untersuchung eingeschlossen. Die Kontrollpopulation bestand aus der KORA-Kohortenstudie, eine repräsentative Bevölkerungsstichprobe aus dem Raum Augsburg, die vom Institut für Epidemiologie des Helmholtz-Zentrums München rekrutiert wurde (Hans-Erich Wichmann). Die statistischen Analysen führte die Arbeitgruppe „Statistische Genetik“ des MPI für Psychiatrie durch (Bertram Müller-Myhsok). Die Untersuchung erfolgte in zwei Stufen. In der ersten explorativen genomweiten Genotypisierungsstufe zeigte sich in sechs unterschiedlichen chromosomalen Regionen eine Assoziation. In der folgenden zweiten Stufe wurden die assoziierten SNPs mit der höchsten Signifikanz in zwei weiteren unabhängigen Patienten- und Kontrollpopulationen aus Deutschland, Österreich und Kanada genotypisiert (Abb. 1 und 2).
.")
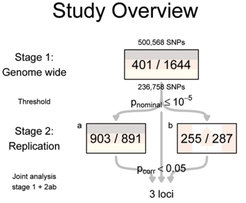
Durch dieses Stufendesign konnten drei genomische Regionen, welche die Gene MEIS1, BTBD9 und MAP2K5/LBXCOR1 kodieren, bestätigt werden. Ein Träger eines dieser drei Risikoallele wird mit einer um 50% erhöhten Wahrscheinlichkeit an RLS erkranken. Homozygote Träger aller drei Risikovarianten weisen ein bis zu 20fach erhöhtes Erkrankungsrisiko auf. Die sog. attributale Risikofraktion der Gene betrug in der deutschen Population 68,6% und in der kanadischen Population 74,2%. Dies bedeutet, dass die genetischen Risikovarianten in diesen drei Genen das Auftreten des Phänotyps zum größten Teil erklären. Insgesamt nahmen über 1600 RLS-Patienten und 2600 Kontrollprobanden an der Untersuchung teil. Forschergruppen aus Deutschland, Österreich und Kanada waren an der Rekrutierung und Phänotypisierung der RLS-Patienten beteiligt.
.")
Bisher ist eine Funktion der identifizierten Gene MEIS1, BTBD9 und LBXCOR1 nur im Zusammenhang mit der embryonalen Entwicklung eines Organismus bekannt. In dieser Phase sind sie an der Ausbildung der Extremitäten und des zentralen Nervensystems beteiligt. Das Homoeoboxgen MEIS1 (Abb. 3) spielt eine Rolle bei der Entstehung der Blutbildung, der Extremitäten und des Neuralrohrs. LBXCOR1 ist ein transkriptionaler Corepressor von LBX. Die Expression von LBX wiederum determiniert eine Gruppe von GABA-ergen Interneuronen im Rückenmark, die an der Verarbeitung von sensiblen Reizen beteiligt sind. Man kann also davon ausgehen, dass eine Verbindung zwischen der sensiblen Komponente des RLS, den Missempfindungen und dem Bewegungsdrang besteht. Welche Funktion diese Gene beim Erwachsenen und insbesondere in Zusammenhang mit der RLS-Erkrankung haben, ist bisher nicht bekannt. Interessanterweise ist das MEIS1-Gen u.a. in der Substantia Nigra exprimiert. Die Zellen dieser Gehirnregion produzieren Dopamin, sodass ein Zusammenhang mit der Wirkung der medikamentösen Behandlung des RLS vermutet werden kann. In einer isländischen Population wurde darüber hinaus eine Assoziation eines Endophänotyps, den periodischen Beinbewegungen, zum BTBD9-Gen gefunden. Dieses Gen spielt daher möglicherweise eine Rolle für die motorische Komponente der Erkrankung.
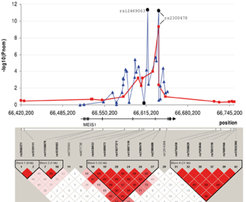
 zeigen klar, dass die RLS-assoziierte Region auf einen einzigen LD-Block innerhalb der transkribierten genomischen Einheit von MEIS1 beschränkt ist. Die Feinkartierung ist in blau dargestellt. Die x-Achse zeigt die genomische Position, die y-Achse den -log 10 des nominellen P-Wertes. Die transkribierte Region des Gens ist durch den schwarzen Pfeil gekennzeichnet, die Exons sind als schwarze senkrechte Balken dargestellt (mit Genehmigung nach [1]).")
Die identifizieren Regionen liegen in intronischen Bereichen der Gene, d.h. in Abschnitten, die nicht direkt in ein Protein übersetzt werden. Die identifizierten Regionen zeigen zwischen verschiedenen Spezies eine sehr hohe Sequenzähnlichkeit. Dieser Umstand weist wiederum darauf hin, dass diese Regionen die Aktivität der Gene, möglicherweise auch ihre Expression beeinflussen.
Zusammenfassend konnten durch die genomweite Assoziationsstudie erstmals genetische Risikovarianten für das RLS-Syndrom identifiziert werden. Dadurch wurde die Voraussetzung sowohl für das Verständnis der molekularen Grundlagen der Erkrankung als auch der bisherigen therapeutischen Ansätze geschaffen. Dieses Wissen wird in Zukunft dazu beitragen, das Ansprechen auf die Therapie genotypabhängig zu untersuchen und eine individualisierte Therapie der Erkrankung zu entwickeln.