Mini-Gehirne aus menschlichen Zellen: Eine neue Technologie zur Erforschung von Fehlbildungen der Großhirnrinde des Menschen
Forschungsbericht (importiert) 2016 - Max Planck Institut für Psychiatrie
Einleitung
Zu den Mechanismen, die der embryonalen Entwicklung des Gehirns zugrunde liegen, gehören die Bildung von Nervenzellen (Neuronen), ihre Migration, das zielgerichtete Wachstum von Nervenzellfasern und die Bildung von Synapsen. In der embryonalen Großhirnrinde, dem Kortex, befinden sich die neuralen Stammzellennahe der Ventrikel, wo sie sich zunächst vermehren und dann neue Nervenzellen bilden (Neurogenese). Neurale Stammzellen sind stark polarisierte Zellen, deren Zellfortsätze sich über die ganze Dicke der Großhirnrinde erstrecken: An einem Ende stehen sie in Kontakt mit dem darunter liegenden Ventrikel und können so auf Signale in der Rückenmarksflüssigkeit (Liquor) reagieren, am anderen Ende stehen sie in Kontakt mit den Hirnhäuten.
 und im Menschen (rechts). Im sich entwickelnden Gehirn entstehen aus am Ventrikel positionierten neuronalen Stammzellen (radialen Gliazellen, grün) die Nervenzellen der Großhirnrinde (blau). Dabei können aus neuronalen Stammzellen durch Zellteilung direkt Nervenzellen gebildet werden, oder über basale Vorläufer- (gelb) und radiale Gliazellen (orange). Im Menschen und anderen Spezies mit Gehirnwindungen ist die Anzahl der basalen radialen Gliazellen deutlich höher als in Arten mit glatter Gehirnoberfläche (wie der Maus) - das ist auch verantwortlich für die höhere Anzahl an Nervenzellen.")

Abb. 1: Schematische Darstellung der Entwicklung des Gehirns in der Maus (links) und im Menschen (rechts). Im sich entwickelnden Gehirn entstehen aus am Ventrikel positionierten neuronalen Stammzellen (radialen Gliazellen, grün) die Nervenzellen der Großhirnrinde (blau). Dabei können aus neuronalen Stammzellen durch Zellteilung direkt Nervenzellen gebildet werden, oder über basale Vorläufer- (gelb) und radiale Gliazellen (orange). Im Menschen und anderen Spezies mit Gehirnwindungen ist die Anzahl der basalen radialen Gliazellen deutlich höher als in Arten mit glatter Gehirnoberfläche (wie der Maus) - das ist auch verantwortlich für die höhere Anzahl an Nervenzellen.
Neurale Stammzellen können nicht nur Nervenzellen generieren, sondern auch noch höher spezialisierte Vorläuferzellen, die basalen radialen Gliazellen [1]. Diese Zellpopulation findet sich besonders häufig bei Spezies mit hochgradig gefalteter Großhirnrinde, wie dem Menschen, was die Vermutung nahelegt, dass sie eine besondere Rolle bei der Vergrößerung des menschlichen zerebralen Kortex spielen [2, 3]. Diese Vorläuferzellen bringen selbst Neurone hervor, die wandern und den zerebralen Kortex bilden, der aus sechs verschiedenen Zellschichten besteht. Jede dieser Schichten enthält eine spezialisierte Population von Nervenzellen (Abb. 1).
Gestörte Entwicklung der Großhirnrinde
Die Bedeutung der spezifischen Anordnung verschiedener Nervenzellen in sechs kortikalen Schichten wird besonders deutlich, wenn die Entwicklung des Großhirns gestört ist: zum Beispiel, wenn die Neurone nicht zu ihrer korrekten Position wandern, sondern an ihrem Entstehungsort nahe der Ventrikel bleiben und so genannte Heterotopien bilden [4, 5]. Fehlbildungen des zerebralen Kortex sind auch das Ergebnis von Beeinträchtigungen der Zellteilung und der Vermehrung von neuralen Stammzellen und führen häufig zu schweren Formen der Epilepsie (Abb. 2).
. Verschiedene Schritte der Entwicklung der Großhirnrinde (obere Zeile) können fehlerhaft verlaufen und zu diversen Entwicklungsstörungen des Gehirns führen (mittlere Zeile). Die häufigsten Folgen sind Entwicklungsverzögerungen, intellektuelle Störungen und Epilepsie.")

Interessanterweise lassen sich die genetischen Ursachen von Fehlbildungen des menschlichen Gehirns in spezifische Klassen unterteilen. Hierzu gehören vor allem Mutationen von Genen, die am Umbau des Zytoskeletts beteiligt sind. Das Zytoskelett ist verantwortlich für die dynamischen morphologischen Veränderungen der Zellen und ihre Bewegung.
Tiermodelle reichen nicht aus
Studien an Mäusen mit Genmutationen, die in Patienten mit kortikalen Fehlbildungen identifiziert wurden, reproduzieren die beim Menschen beobachteten Erkrankungen nur teilweise. Somit sind Tiermodelle nicht ausreichend, um die für diese Erkrankungen verantwortlichen Mechanismen zu erklären [4, 5]. Einige Mauslinien bilden die Fehlbildungen des zerebralen Kortex zwar umfassend ab [6] und ermöglichen eine tiefergehende Charakterisierung der molekularen und zellulären Mechanismen, die der Entwicklung dieser Krankheiten zugrunde liegen (Abb. 1). Dennoch zeigen die umfassenden Unterschiede zwischen den Phänotypen von Menschen und Mäusen, wie extrem komplex und divers der zerebrale Kortex organisiert ist. Für die Forschungsgruppe ist es daher entscheidend, alternative Modelle zu nutzen, welche die Untersuchung des Verhaltens menschlicher Zellen ermöglichen.
Es ist entscheidend, kortikale Fehlbildungen in Mausmodellen und parallel in menschlichen Modellen abzubilden, wenn man seit Langem bestehende Fragen beantworten will: Etwa die Frage, welche gemeinsamen oder artspezifischen molekularen und zellulären Mechanismen der Entwicklung kortikaler Fehlbildungen zugrunde liegen, oder die Frage, auf welche Weise heterotopische Nervenzellen die neuronalen Schaltkreise stören und so zur Epilepsie führen.
Heterotopien sind häufig auf bestimmte Hirnareale beschränkt, was darauf schließen lässt, dass nicht alle Vorläuferzellen und Neurone betroffen sind. In Zusammenarbeit mit Ärzten wurde eine Exom-Analyse an Patienten durchgeführt, die an Heterotopien leiden. Bei dieser Analyse wurden die für Proteine kodierenden Bereiche der DNA von Patienten und ihren gesunden Eltern sequenziert und miteinander verglichen. Die Untersuchung brachte neue Genmutationen ans Licht.
Analyse von humanen neuralen Stammzellen und Neuronen
Vor einigen Jahren entdeckten Yamanaka und seine Kollegen eine Reihe von Transkriptionsfaktoren, die den zellulären Phänotyp einer Körperzelle in den einer pluripotenten Stammzelle zurückverwandeln können. Da man sie mit bestimmten Faktoren zu verschiedenen Zelltypen differenzieren kann, bieten diese induzierten pluripotenten Stammzellen (iPS-Zellen) in nie dagewesener Weise die Möglichkeit, spezifische Modelle zur Untersuchung von Krankheitsbildern beim Menschen auf unterschiedlichem genetischem Hintergrund zu entwickeln. Außerdem können mit ihrer Hilfe neue Medikamente zur Therapie entdeckt und getestet werden. Die Möglichkeit der zellulären Reprogrammierung von Körperzellen hat das Modellieren menschlicher Erkrankungen und Fehlbildungen revolutioniert [7]. In kürzester Zeit haben sich reprogrammierte Zelllinien zu einer unverzichtbaren und flexiblen zellulären Plattform sowohl für die Grundlagenforschung als auch für die regenerative Medizin entwickelt. So konnte ein passendes Modell zur Entwicklung des menschlichen Gehirns etabliert werden, das die einzigartige Möglichkeit bietet, Modellsysteme von Fehlbildungen zu validieren und sich der Komplexität des menschlichen Gehirns anzunähern (Abb. 3).
 reprogrammiert werden. Diese werden zu neuronalen Stammzellen und Nervenzellen differenziert (separat oder in Form von komplexeren Rosetten). Des Weiteren können zerebrale Organoide aus iPSCs gewonnen werden - das beste momentan verfügbare Modellsystem für das sich entwickelnde menschliche Gehirn. Die unterste Zeile zeigt Beispielbilder für die jeweiligen Schritte.")
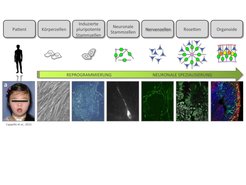
Abb. 3: Schema zur Herstellung der humanen 2D und 3D Modellsysteme. Von Patienten mit Entwicklungsstörungen des Gehirns können Körperzellen entnommen und zu induzierten pluripotenten Stammzellen (iPSCs) reprogrammiert werden. Diese werden zu neuronalen Stammzellen und Nervenzellen differenziert (separat oder in Form von komplexeren Rosetten). Des Weiteren können zerebrale Organoide aus iPSCs gewonnen werden - das beste momentan verfügbare Modellsystem für das sich entwickelnde menschliche Gehirn. Die unterste Zeile zeigt Beispielbilder für die jeweiligen Schritte.
Menschliche IPS-Zellen werden aus den Körperzellen von Patienten generiert und können in neuronale Stammzellen und Neurone differenziert werden. Mit Hilfe des CRISPR/Cas9-Systems, einer kürzlich entwickelten Technologie zum Genome-Editing, können Kontroll- und Patientenzelllinien mit demselben genetischen Hintergrund hergestellt werden, die sich nur durch Mutationen in bestimmten Genen unterscheiden. So kann die Rolle dieser spezifischen Gene in der Entwicklung des Gehirns untersucht werden. Dadurch lassen sich allgemeine zelluläre Mechanismen identifizieren, die der Bildung von Heterotopien zugrunde liegen. Außerdem können die zellulären und funktionalen Eigenschaften der umprogrammierten neuronalen Stammzellen und Neurone untersucht und so verschiedene Entwicklungsprozesse eingehend unter die Lupe genommen werden. Das endgültige Ziel ist es, die elektrophysiologischen Eigenschaften von Nervenzellen innerhalb des menschlichen Zellmilieus zu analysieren.
“Mini-Gehirne“ liefern neue Erkenntnisse
Gleichzeitig können aus Patientenzellen zerebrale Organoide oder „Mini-Gehirne“ als dreidimensionale Strukturen entwickelt werden: Die Zellen werden zunächst in pluripotente Stammzellen reprogrammiert. Aus diesen Stammzellen entwickeln sich in vitro komplexe 3D-Strukturen, die Gehirn-spezifische Zelltypen enthalten. Die so entstandenen Zellverbände spiegeln in einer sehr vereinfachten Form die Gehirnentwicklung, speziell des Gehirns des Patienten, wider. Aufgrund ihrer einfachen Gewinnung und der Ähnlichkeit mit menschlichen Organen öffnen Organoide neue Möglichkeiten für die translationale Forschung, der weitestgehend direkten Übertragung von Forschungserkenntnissen in die klinische Anwendung. Zerebrale Organoide oder Mini-Gehirne gelten als vielversprechend für die regenerative Medizin. Sie sollen Medikamententests erleichtern und neue Erkenntnisse über die molekularen und zellulären Eigenschaften des Gehirns liefern.
Im Hinblick auf die oben beschriebenen Fragestellungen können diese Technologien dazu beitragen, die Wissenslücke zu schließen, die zwischen den Resultaten aus Mausmodellen und dem Wissen über Fehlbildungen im menschlichen Gehirn klafft. Die Identifikation eines Netzwerks von Genen und Signalwegen, die für kortikale Fehlbildungen verantwortlich sind, wird - zusammen mit dem Verständnis für die funktionalen Aspekte dieser Krankheiten - wegweisend sein bei der Suche nach gemeinsamen molekularen und zellulären Mechanismen. Das kann dazu beitragen, neue Strategien für einen therapeutischen Ansatz zu entwickeln. Das übergeordnete Ziel der Max-Planck Forschungsgruppe ist es, Fehlbildungen zu korrigieren und ein diagnostisches, genetisches Screening zu entwickeln, das sehr frühzeitig die Patienten identifiziert, die das Risiko in sich tragen, eine Epilepsie zu entwickeln.
Literaturhinweise
Neurobioloogy of Diseases 38, 154-166 (2010)
Annual Review of Cell Developmental Biology 20, 593-618 (2004)
Frontiers in Cellular Neuroscience 9, 30 (2015)